
Primary Hyperoxaluria
Focus Group
Dawn S. Milliner, MD. (Principal Investigator) | bio
| Mayo Clinic, Rochester, MN
Julie Olson, RN (Study Coordinator) | Mayo Clinic, Rochester, MN
Barbara M Seide (Study Coordinator) | Mayo Clinic, Rochester, MN
Alicia Meek (Study Coordinator) | Mayo Clinic, Rochester, MN
Contact Information:
Mailing address for records/paperwork:
Mayo Clinic
200 First Street SW
Rochester, MN 55905
Mayo Clinic Hyperoxaluria Center Ei-SL 33
Email: hyperoxaluriacenter@mayo.edu
Phone: 800-270-4637
Fax: 507-255-0770
Facebook:
http://www.facebook.com/hyperoxaluria
What's New?
Save the Date:
RKSC meeting ASN 2013
The RKSC will host their annual meeting at the 2013 ASN in Atlanta, Georgia on Saturday, November 9th at 12:45-1:45 EST. For additional details contact Tammy Evans: evans.tamara@mayo.edu
11th International Primary Hyperoxaluria Workshop & Patient Meeting
Chicago, IL
June 27-29, 2014
- Details to follow
2013 OHF Patient Day Meeting at Mayo Clinic

June 8, 2013 RKSC and OHF (Oxalosis and Hyperoxaluria Foundation) hosted a PH and Dent disease Patient Day at the Mayo Clinic in Rochester, MN. The meeting included presentations on both diseases by Mayo Clinic physicians and OHF reported on the importance of Patient Advocacy Groups being involved with the RKSC and also with research and not only fund raising but also with disease awareness.
After the meeting we had a very successful turn out for the "OHF Walk for Kidneys”! Everyone had a great time.
Advocacy Organization
Oxalosis and Hyperoxaluria Foundation | www.ohf.org
Disease Information

Expand All Topics | Close All Topics
The primary hyperoxalurias are autosomal recessive disorders of glyoxylate metabolism characterized by excessive production and urinary excretion of oxalate.
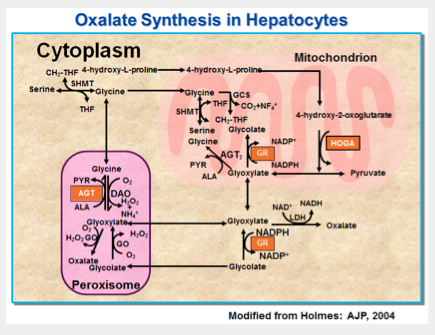
The glyoxylate pathway in the human hepatocyteAlanine:glyoxylate aminotransferase (AGT). Glyoxylate reductase: hydroxypyruvate reductase (GR/HPR). Glycolate oxidase (GO). Lactate dehydrogenase (LDH). D-amino acid oxidase (DAO). Pyridoxal phosphate (PLP). Deficiency or mistargeting of AGT (PHI) results in buildup of glyoxylate and increased oxalate production. Deficiency of a protein with dual GR and HPR activities (PHII) gives rise to increased hydroxypyruvate and glyoxylate, precursors of L-glycerate and oxalate, respectively.
The term "primary hyperoxaluria" was first used by Archer and colleagues in 1957 to specifically denote a suspected metabolic origin for the marked hyperoxaluria, recurrent urolithiasis and renal and extra-renal calcium oxalate crystal deposition that characterized affected children. The urine oxalate excretion rate in affected patients is typically 3 to 6 times normal with severe clinical consequences. Kidney stones and/or calcification of the kidney are most often evident in childhood or adolescence, since the hyperoxaluria is present from birth. However, in some patients, symptoms are not recognized until early or even mid adulthood. . Renal injury due to oxalate and consequences of the stones often leads to renal failure. Loss of kidney function, if not addressed promptly by transplantation, leads to markedly increased plasma concentrations of oxalate resulting in deposition of calcium oxalate in body tissues (oxalosis). Resulting organ system dysfunction including ischemic ulcers of the skin, metabolic bone disease, refractory anemia, cardiomyopathy, and cardiac conduction system abnormalities are the cause of severe morbidity and mortality. Historically, the median age at death was only 36 years. With advances in diagnosis and treatment, much better outcomes are now achievable.
PH Type
There are three forms of primary hyperoxaluria for which the specific genetic cause has been confirmed.
- Type 1 is caused by mutations of the AGXT gene. These mutations result in a deficiency of the enzyme alanine glyoxylate aminotransferase (AGT) which is found only in the liver.
- Type 2 is caused by mutations of the GRHPR gene. These mutations result in a deficiency of the enzyme glyoxylate reductase/hydroxypyruvate reductase (GR/HPR) found in the liver and other tissues.
- Type 3 is caused by mutations of the HOGA1 gene (formerly DHDPSL) which result in deficiency of the enzyme 4-hydroxy-2-oxoglutarate aldolase found in the liver. Though deficiency of this enzyme has been confirmed in PH3, understanding of the mechanisms whereby this leads to oxalate overproduction are unclear.
The metabolic pathways involved, all present in the hepatocyte, are shown in Fig 1. Pyridoxine (vitamin B6) is a cofactor for the enzyme AGT, deficiencies of which are the cause of PH1. Administration of pharmacologic doses of this vitamin can reduce urine oxalate levels in PH1 patients with certain genotypes. Pyridoxine is not effective in all PH1 patients, and is not effective in patients with PH2 or PH3. Additional types of primary hyperoxaluria remain to be identified. Patients with similar degrees of hyperoxaluria and clinical characteristics who lack demonstrable mutations of AGXT, GRHPR, and HOGA1 have been well described, and are considered to have unclassified primary hyperoxaluria. Differences in outcome among the three types of PH are beginning to be understood. PH1 has the most severe manifestations. Untreated, patient outcome in PH1 is often poor, with death from renal failure and systemic oxalosis the norm. However, there is wide variability in outcome among patients, and with active ongoing treatment patient survival with preserved renal function to middle age (or older) is possible. Patients with PH2 and PH3 appear to have milder disease. They demonstrate much better preservation of kidney function and fewer stones. The important factors that influence frequency and severity of stone formation, kidney function, and patient survival remain poorly understood.
In all cases, care by a physician who is familiar with treatment of PH can be instrumental in reducing adverse effects of the hyperoxaluria. Treatment strategies may include maximizing oral fluid intake, dietary modification, pharmacologic doses of pyridoxine for those patients in whom it is effective, and neutral phosphate and/or citrate to reduce urinary supersaturation with calcium oxalate. Renal function must be monitored vigilantly and renal replacement therapy should be initiated promptly if renal clearance falls below a critical threshold, in order to prevent body-wide deposition of calcium oxalate (systemic oxalosis). Standard dialysis regimens are unable in most PH patients to remove sufficient amounts of oxalate to prevent systemic oxalosis. Kidney transplantation alone or combined kidney-liver transplantation is the preferred form of renal replacement therapy for PH patients. If kidney function declines, and while awaiting transplantation, patients must be aggressively dialyzed, often 6 or 7 days per week and/or using a combination of modalities, in order to remove enough oxalate to prevent body-wide oxalosis and its severe consequences.
Clinical Aspects
Primary hyperoxaluria (PH) is the most severe of the hereditary causes of nephrolithiasis. Enzyme deficiency in the liver results in marked overproduction of oxalate that must be excreted by the kidneys. Oxalate in the urine in high concentrations is poorly insoluble and when combined with calcium thus leads to calcium oxalate crystals and stones.
Urinary tract stones cause pain, and urologic procedures are often required for their removal. Calcium oxalate crystals are also found in the parenchyma of the kidney. Damage to surrounding tissue over time results in renal failure in the majority of patients with type 1 PH, and a minority of patients with type 2 disease. Most PH3 patients studied thus far have maintained satisfactory kidney function. As kidney function declines and excess oxalate can no longer be eliminated effectively, blood oxalate concentrations rise, and calcium oxalate crystals deposit in multiple organs and tissues (systemic oxalosis). Calcium oxalate in bone results in fracturing bone disease as well as erythropoietin resistant anemia due to replacement of marrow by oxalate. Painful ischemic ulcers of extremities occur due to involvement of blood vessels, cardiomyopathy due to deposition in myocardium, and fatal cardiac arrhythmias are the consequence of deposition in the cardiac conduction system, among other problems. In the majority of patients with type 1 PH, kidney transplantation alone is not sufficient, since the transplanted kidney is damaged by ongoing high oxalate production. Liver and kidney transplantation must be performed. Among patients with PH 1, the median age at kidney failure is 33 years of age. Disease expression, however, varies widely. Some PH patients progress to end stage kidney failure within the first year of life, while others maintain satisfactory kidney function until well into middle age. This variability has not been satisfactorily explained and is only partially influenced by genotype or other factors such as degree of hyperoxaluria. Modifiers of disease expression, if identified, offer promise as potential new strategies for treatment.
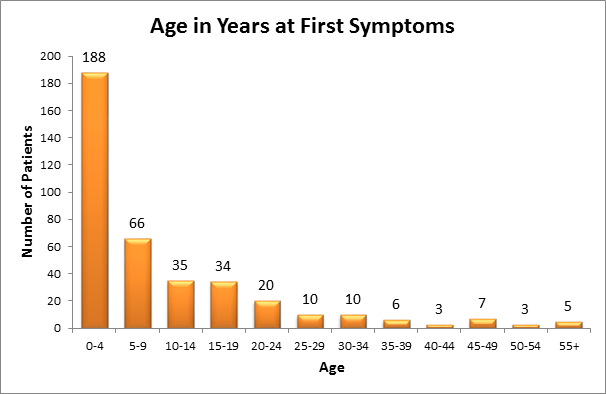
Though hyperoxaluria is present from birth, symptoms may first appear any time from infancy to adulthood, and symptoms can range from mild to severe. The first sign of primary hyperoxaluria is most often kidney stones with associated symptoms. Because kidney stones are uncommon in childhood, all children and adolescents who are found to have calcium containing kidney stones should be screened for hyperoxaluria. Pediatric patients with hyperoxaluria can then be screened for primary hyperoxaluria by means of more specialized testing.
Primary hyperoxaluria that remains untreated may result in kidney failure, and if not dialyzed aggressively or the recipient of a transplant may lead to systemic deposition of oxalate (oxalosis). Sometimes, a patient may have already progressed to oxalosis and, during an eye examination, his or her ophthalmologist may discover oxalate crystals in the patient's eyes. Oxalosis in its late stages will cause bone disease, the result of oxalate crystals depositing in the bones and joints. It may also cause anemia that is difficult to treat, skin ulcers, and heart problems. The resulting morbidity can be a cause of death.
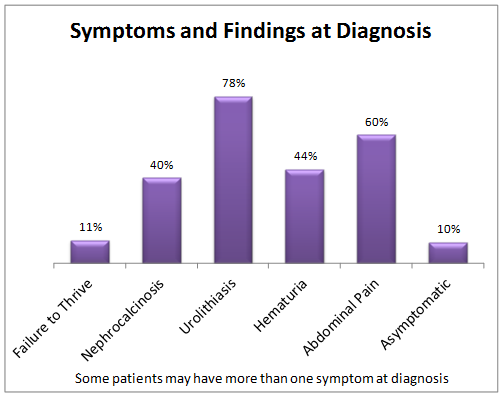
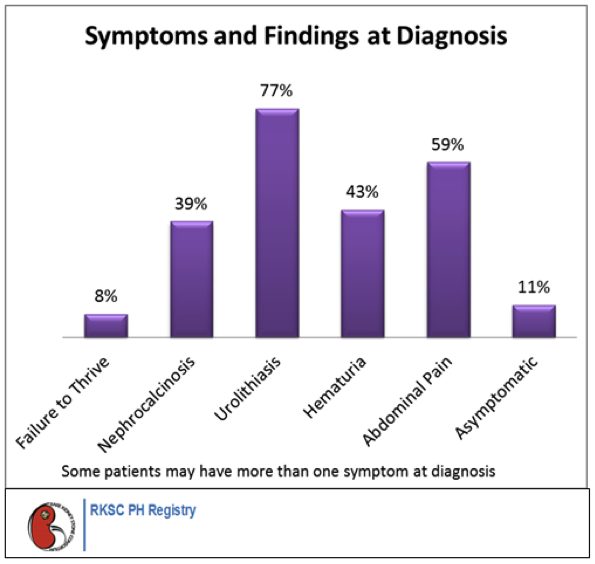
All the pathological sequelae of the primary hyperoxalurias are related to the increased synthesis and excretion of oxalate. Marked hyperoxaluria is present from birth on, with 2 to 8 times the upper limit of normal urine oxalate being characteristic. Blood in the urine or pain related to stones, stone passage, or urinary tract infection are the most common symptoms of the disease. Over time, frequent stone recurrences and the need for multiple stone removal procedures are typical. The majority of patients are symptomatic before 10 years of age. In some cases, however, the disease may go unrecognized either due to the absence of symptoms or to incorrect diagnosis, until patients reach 30 to 50 years of age. Oxalate at high concentrations combines with calcium in the urine to form crystals in the urinary tract leading to stones, and also deposit in kidney tissue causing nephrocalcinosis. Calcium oxalate crystals are injurious to renal cells and incite a granulomatous reaction in the renal interstitium. Over time the effects of such injury, often combined with intermittent obstruction or infection related to stones, lead to kidney failure.
Earlier literature showed that about 50% of patients developed kidney failure by 15 years of age, and about80% developed kidney failure by age 30. With improved diagnosis and management, more recent information shows the median age at renal failure to be 33 years. However, some patients present with kidney failure as the first manifestation of the disease, as early as 4 months of age. Patients with type 2 and type 3 disease appear to have a milder course overall than those with type 1, including better preservation of renal function. The reasons for such variation in clinical expression are poorly understood, and if elucidated may provide valuable insights as to potentially remediable factors that can be exploited for therapeutic benefit. Early diagnosis of primary hyperoxaluria is of vital importance so that treatment can be initiated as soon as possible. Yet, due to lack of familiarity with the disease, delays of many years from onset of symptoms to diagnosis are common. Among patients in the RKSC Primary Hyperoxaluria Registry, the mean time from symptom onset to diagnosis was 5.6 years (0.0, 6.5 25th, 75th%ile). Nineteen percent of patients were first recognized to have PH only after developing irreversible renal failure.
Patients with hyperoxaluria or suspected PH should have a comprehensive physical exam, including a medical history, family history, and diet and medication history. Diagnostic testing to confirm hyperoxaluria of a degree consistent with PH (generally > 0.8 mmol/1.73m2/24 hours in a patient with good kidney function), and urine testing to suggest abnormalities of the glyoxylate metabolic pathway (urine glycolate, glycerate, glyoxylate and hydroxyoxoglutarate) can be helpful., Any patient with reduced kidney function and nephrocalcinosis or stones should be checked for hyperoxalemia as well as hyperoxaluria. A careful physical examination, ophthalmologic examination for retinal crystals, and an ECG and echocardiogram for oxalate cardiomyopathy or conduction disturbances, can be helpful. X-rays of the long bones and bone density testing may provide clues to oxalate osteodystrophy.
Definitive diagnosis is most often by DNA screening for mutations of AGXT, GRHPR, and HOGA1. Occasionally a liver biopsy may be needed to confirm enzyme deficiency. An algorithm for diagnosis of PH was published in 2012 (Pediatric Nephrology).
The only definitive treatment for type 1 PH is liver transplantation to replace the missing AGT enzyme. However, complete removal of the patient's otherwise normal liver is required in order to eliminate oxalate overproduction. Risks of the transplant procedure, as well as the risks of life-long immune suppression. In 30-50% of PH1 patients, reduction in urine oxalate excretion can be achieved by treatment with pharmacologic doses of pyridoxine, suggesting enhancement of AGT enzyme activity. This effect is specific to certain mutations of AGXT. The pyridoxine effect is partial when there is heterozygosity for these mutations, but normalization of urine oxalate has been described with homozygosity (Monico article). AGT requires pyridoxal phosphate as a co-factor. Though the mechanism of its effect in PH1 patients remains to be proven, in vitro studies demonstrate molecular stabilization of mutant enzyme by chaperones such as pyridoxal phosphate and betaine.
In patients with all forms of PH, treatments directed to reduction in crystal and stone formation are of value and include high oral fluid intake to reduce urine oxalate concentration, and oral medications that inhibit calcium oxalate crystal formation, specifically citrate or phosphate. Though long term outcomes have improved with earlier diagnosis and currently available treatment, renal failure nonetheless still develops in 70% of patients with PH type 1 by 60 years of age. More effective treatments are urgently needed. Promising new directions using molecular chaperones, oxalate degrading bacteria, and exploitation of oxalate transport physiology are in various stages of investigation.
Treatment should be individualized for PH patients, taking into account each patient's needs, the type and severity of disease and how well the patient responds to treatment. Patients will need to be closely monitored by their medical team to measure their responses to the following treatments:
Diet modifications
Dietary restrictions are not as important in primary hyperoxaluria, since the source of the excess oxalate is metabolic production. There is no evidence that dietary oxalate restriction is of value. However, avoidance of dietary oxalate excess seems prudent. A dietician can assist with diet modifications and recommendations.
High fluid intake
Patients with hyperoxaluria who do not have kidney failure should maintain high oral fluid intake at all times. In occasional circumstances fluid intake by gastrostomy or other enteral administration is indicated. This may occur in infants or young children where compliance with high oral fluid is difficult to maintain.
Medications
All patients with primary hyperoxaluria type 1 or PH of unknown type should receive prescription-level doses of pyridoxine (Vitamin B6) of 5-8 mg/kg/day for at least a 3 month trial period. Neutral phosphates or citrate are also effective in reducing calcium oxalate crystals and stones in the urine due to their direct inhibitory effect on calcium oxalate crystal formation.
Kidney Stone Management
Large kidney stones that cause pain or other symptoms or obstruct the flow of urine from the kidneys may require removal or fragmentation. A urologist experienced in stone removal procedures should be consulted to recommend the best approach.
Clinical Studies
New study open for enrollment: Influence of Hydroxyproline Plasma Concentration on its Metabolism to Oxalate. Oral intake of hydroxyproline leads to increased urinary excretion of oxalate in normal subjects. This study will enroll patients who are known to have primary hyperoxaluria and examine how hydroxyproline is broken down in a PH patient's body. The study will help us determine if hydroxyproline is metabolized differently in patients with PH in healthy controls.
There will be 24 participants enrolled in this study:
6 patients in each group: PH type 1 = 6 patients
PH type 2 = 6 patients
PH type 3 = 6 patients
PH Non 1/Non 2/Non 3 = 6 patients
The study will be done at Mayo Clinic's Clinical Research Unit in Rochester, MN.
- Patients will have two phone appointments with a Mayo Clinic dietician to dicuss detailed diet instructions. This diet, which uses normal foods, will be followed for sevreral days before coming to the Clinical Research Unit.
- Patients will receive an IV infusion of hydroxyproline over 6 hours in the Clinical Research Unit
- Blood and urine samples will be taken before, during and after the infusion in order to check the patient's levels of hydroxyproline, oxalate and glycolate
For more information on this study contact our Study Coordinators at: hyperoxaluriacenter@mayo.edu or 800-270-4637
Additional PH Studies open for enrollment:
RKSC Primary Hyperoxaluria Registry
The Rare Kidney Stone Consortium has opened enrollment for the Primary Hyperoxaluria Registry. In this registry, patients with PH will be enrolled and information collected about their condition annually. The collected data in this registry will help provide a better understanding of the condition. The goal of this registry is to collect data about this rare disease, that may help to develop better treatment protocols in the future. Active enrollment of patients continues. While we have made good progress in the PH Registry enrollment, we are anxious to increase our enrollment and we invite all physicians to help! If you have any PH patients who might be interested in participating, please contact one of our Study Coordinators and they will make this possible: hyperoxaluriacenter@mayo.edu or 800-270-4637
To see some of what we are learning from the PH Registry click here
Correlation of Disease Expression with Specific Genetic Mutations in Primary Hyperoxaluria
This protocol will demonstrate that certain mutations predispose individuals with hyperoxaluria to more severe disease.
Oxalate excretion is in part genetically determined. To date, mutations in two separate loci are known to cause hyperoxaluria. Primary hyperoxaluria types 1 (OMIM 259900) and 2 (OMIM 260000), result from mutations in AGXT (2q36-q37) and GR/HPR (9q13-q21), respectively. An influence from other genetic loci is suspected, however, based on a wide phenotypic spectrum of disease expression. To date, over 100 mutations have been catalogued in AGXT and GR-HPR. Different mutations appear to have widely differing effects on AGT and GR-HPR protein quantity and activity. Attempts to correlate specific genetic mutations with disease phenotype have been disappointing, however. There are also many polymorphisms in both genes, some of which are functional, further complicating correlations between genotype and phenotype.. Because many mutations are private or family-specific, segregation analysis is often needed, requiring genotyping in family members.
Investigations into the Genotype and Phenotype of Unclassified Hyperoxaluria: Enteric Oxalate Absorption Study
This study will measure urinary oxalate excretion using 13C-labelled oral oxalate loads. Flavored gelatin (children) or a capsule (adults or children) containing 13C-labelled (a stable isotope) oxalic acid will be ingested by each subject. Urine specimens will be analyzed for 13C oxalate by gas chromatography/mass spectroscopy (GC/MS). The utility of using urine oxalate as an indicator of oxalate absorption is based on the finding that > 90% of an absorbed dose of radioisotope labeled oxalate can be recovered from the urine in 24-36 hours. Most of the oxalate excretion after administration of 13C-labelled oxalate occurs within the first 6 hours after ingestions. We anticipate that the cause of oxalate excess in the patients is due to either intestinal factors or secondary to metabolic overproduction. The degree of hyperoxaluria that has generally been observed in patients with malabsorption states is in the range of < 1 mmol/1.73 m2/24 hrs (oxalate excretion rate in normal adults is between 0.11 and 0.4 mmol/1.73m2/24 hrs via the oxalate oxidase method). Based on this information, and the marked hyperoxaluria of greater than 1 mmol/1.73m2/24 hrs, we anticipate enteric oxalate absorption to be normal in patients with unclassified primary hyperoxaluria.
For additional study information or to participate in the above studies:
Contact the study coordinator by phone or by e-mail.
Contact: Mayo Hyperoxaluria Center Study Coordinators: Julie Olson, Barb Seide, Alicia Meek
Phone: 800-270-4637
Email: hyperoxaluriacenter@mayo.edu
Basic Research
Ongoing Primary Hyperoxaluria research includes multidisciplinary collaboration with investigators in Nephrology, Urology, Radiology, Laboratory Medicine and the basic sciences:
- Determination of specific genetic mutations in primary hyperoxaluria patients, and correlation with disease outcomes
- Development of newer radiology techniques in order to measure renal calcium content and stones
- Development of better rat models of hyperoxaluria
- Study of the mechanism(s) by which oxalate changes cell function
- Development of a Tissue Bank for urine, plasma, whole blood and liver samples collected from patients to facilitate investigation and collaborative research.
REGISTRY MISSION STATEMENT
The Rare Kidney Stone Consortium PH Registry is a vehicle through which the international scientific community can pool information regarding these rare diseases, in order to collectively advance knowledge, and ultimately improve the quality of life of affected individuals and their families.
The Registry Database Study design is that of an observational cohort, following patients forward in time from the date of enrollment in the Registry. Limited data will also be collected regarding patient status at time of diagnosis, which may be several years earlier. It is expected that patients will be evaluated approximately annually, though this cannot be mandated. As enrollment in the Registry is voluntary, it will not be possible to estimate disease prevalence or incidence. However, it is hoped that sufficient numbers of representative patients will be enrolled to allow us to characterize the disease(s) and describe its natural history.
Registry date can be collected and entered either by physician participation in the Registry or by patient cooperation with Registry staff. Physicians engaged in the management of patients with PH complete a web-based on-line enrollment form with information on their name, specialty, type of practice, and contact information. In order to enter patients the participating physician will be responsible for securing Institutional Review Board (IRB) or Independent Ethics Committee (IEC) approval for the research, and a patient authorization form if required by the local IRB. To help in this, a sample Patient Authorization request form will be submitted to the physician upon receipt of physician enrollment. A copy of each signed research authorization, if required by the local IRB, should be retained at the enrolling institution. After the approvals are obtained, physicians may enroll patients into the Registry by completing secure web based patient enrollment forms. After a patient is enrolled, the referring physician will be sent an invitation letter for the patient to participate in other activities of the Registry. If the patient consents, kits will be sent for blood and urine samples, as appropriate. A patient who does not have a physician willing to complete the necessary steps to enrollment can participate by providing his/her medical records to the Mayo Clinic staff, and signing a consent for Registry participation. Mayo staff will then enter the medical information in the Registry. Patient enrollment forms are completed by an enrolled physician or his/her designated staff following patient authorization and IRB approval, as required by the physician's site. These have been converted to a Web-based format. A short initial form is completed for each patient to confirm eligibility. If the patient meets eligibility requirements, a unique study number is assigned and a registration form is completed to include the patient's authorization of participation and demographic information. Thereafter, the patient is identified only by his/her unique alphanumeric code. An initial enrollment form for each patient establishes specific findings at diagnosis, and key features of the clinical course to that date. A current status form is then completed. The current status form will be updated annually. A confirmation of patient enrollment is sent on satisfactory receipt of patient enrollment form and will include a unique Registry identification number, coded patient initials, gender and date of birth. For each subject, a unique identification number is provided by the Registry. Following assignment of the unique patient identifier (letters and number), in order to best protect patient confidentiality, only this identifier will be used in subsequent communications with the physician or study center.
Your participation is very important for building a database of information to assist in developing therapeutic strategies for Primary Hyperoxaluria. It will help to outline more about disease expression, and also about diagnosis, therapy, transplantation procedures and outcomes. A study coordinator is available to answer questions or help in any way with data entry. To contact the study coordinator please call (800) 270-4637 OR E-mail to hyperoxaluriacenter@mayo.edu
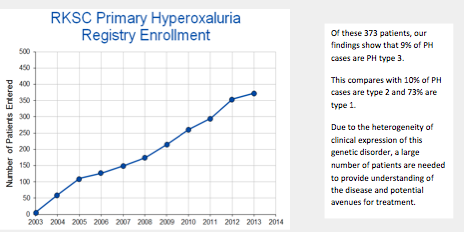
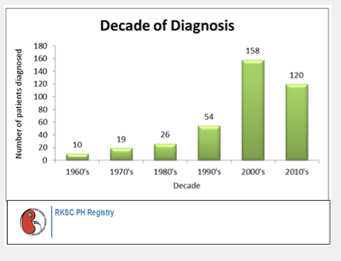
RKSC Registry as of July 2013
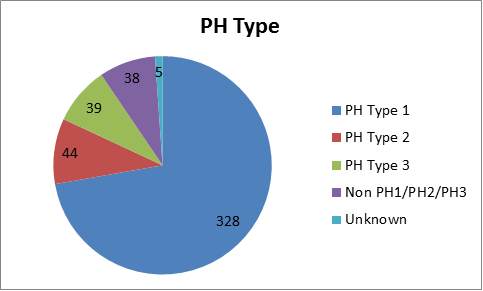
The PH Registry continues to be an important tool in PH research. Thanks to the PH community's help and participation, there are now 373 patients enrolled in the Primary Hyperoxaluria Registry.
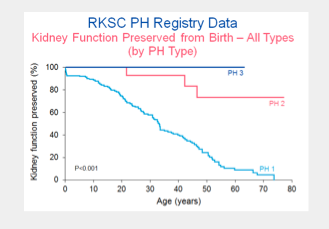
The PH Registry has been valuable in understanding the implications of PH type in renal outcomes. Maintenance of kidney function from birth through age 80 years is shown below for primary hyperoxaluria types 1,2 and 3.
Active enrollment of patients continues. While we have made good progress in the PH Registry enrollment, we are anxious to increase our enrollment and we invite all physicians to help!
If you have any PH patients who might be interested in participating, please contact one of our Study Coordinators and they will make this possible.
PH Type
There are currently at least 3 types of defined of primary hyperoxaluria.
Other patients with similar clinical features appear to have a PH of undefined cause.
- Type 1 is caused by mutations of the AGXT gene. These mutations result in a deficiency of the enzyme alanine glyoxylate transferase (AGT) which is found only in the liver.
- Type 2 is caused by mutations of the GRHPR gene. These mutations result in a deficiency of the enzyme glyoxylate reductase/hydroxypyruvate reductase (GR/HPR) found in the liver and other tissues.
- Type 3 is caused by mutations of the HOGA1 gene (formerly DHDPSL) found in the liver.
RKSC Registry as of July 2013

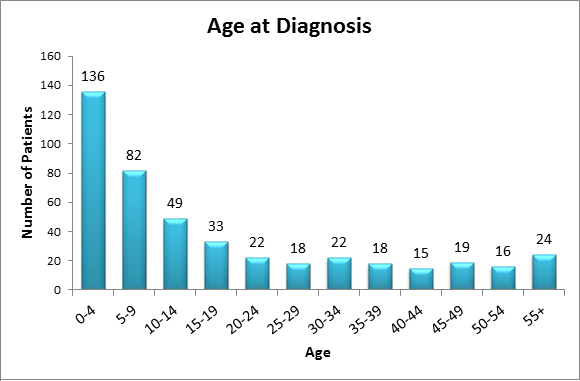
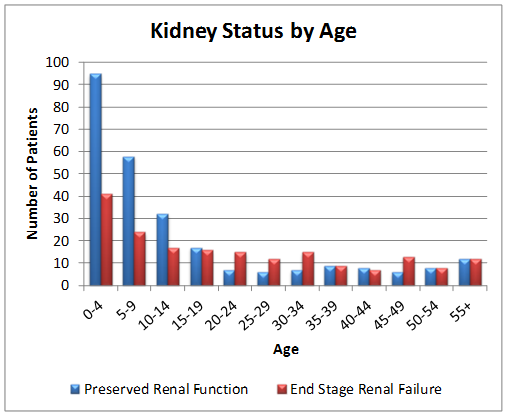
- Mayo Clinic Hyperoxaluria Center is a resource for patients and their families and physicians. The center facilitates collaborative research to provide better understanding of this disorder.
- Contact us at: hyperoxaluriacenter@mayo.edu or call 800-270-4637
- Oxalosis and Hyperoxaluria Foundation (OHF)
This is the only organization in the world dedicated to improving the care and treatment and finding a cure for Oxalosis, PH and related stone diseases. Website: www.ohf.org or 800-643-8699 - Rare Kidney Stone Consortium PH Registry
The purpose of this registry is to identify worldwide as many affected individuals as possible, and to collect as much clinical information about these patients as is feasible. - The Mayo Clinic Hyperoxaluria Center is a resource for physicians treating or diagnosing PH patients. The PH Center staff can assist with recommendations for specialized testing for PH including genetic testing that is available thru Mayo Medical Laboratories (MML): http://www.mayomedicallaboratories.com/
- The Mayo PH Center physicians are available for assistance with interpretation of PH testing.
MML's Communique Newsletter and PH specific testing available:
http://www.mayomedicallaboratories.com/articles/communique/2013/07-hyperoxaluria-oxalosis/index.html
New algorithms for diagnosis
The RKSC investigators have published a new article that includes updated diagnostic algorithms for the four rare kidney stone diseases in the RKSC, including PH.
Edvardsson V, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani L. Milliner DS, and Palsson R. Hereditary Causes of Kidney Stones and Chronic Kidney Disease. Ped Neph 2013.<(
Article link: http://rd.springer.com/article/10.1007/s00467-012-2329-z
References
Edvardsson V, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani L. Milliner DS, and Palsson R. Hereditary Causes of Kidney Stones and Chronic Kidney Disease. Ped Neph 2013.
Recent publications:
Worcester E, Evan E, Coe F, Lingeman J, Krambeck A, Sommers A, Phillips C, Milliner D. A test of the hypothesis that oxalate secretion produces proximal tubule crystallization in primary hyperoxaluria type 1. Am J Physiol Renal Physiol. Oct 2013
Edvardsson V, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani L. Milliner DS, and Palsson R. Hereditary Causes of Kidney Stones and Chronic Kidney Disease. Ped Neph 2013.
Riedel TJ, Knight J, Murray MS, Milliner DS, Holmes RP, Lowther WT. 4-Hydroxy-2-oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. Biochim Biophys Acta. 2012 Oct; 1822:1544-52.
Beara-Lasic L, Edvardsson VO, Palsson R, Lieske JC, Goldfarb DS, Milliner DS. Genetic causes of kidney stones and kidney failure. Clin Rev Bone Min Metab 2012; 10:2-18.
Monico CG, Milliner DS. Genetic determinants of urolithiasis. Nat Rev Nephrol. 2012; 8:151-62.
Monico C, Rossetti S, Belostotsky R, Cogal A, Herges R, Seide B, Olson J, Bergstrahl E, Williams H, Haley W, Frishberg Y, Milliner D. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol 6:2289-2295, 2011.
 Consortium Home
Consortium Home Primary Hyperoxaluria
Primary Hyperoxaluria